Ana Flávia Arantes Pereira 1
Liege Aparecida Mapa 1
Luna Elisabeth Carvalho Ferreira 1
Nathália Martins Moreira 1
Paula Guimarães Chiesa 1
Sonaly Cristine Leal 2
George Schayer Sabino 2
Anderson Hollerbach Klier 3
.
Resumo: Com o intuito de facilitar a aprendizagem e minimizar as dificuldades no entendimento de parâmetros químicos conformacionais, foi proposta a construção virtual de uma série de estruturas carbônicas, tanto homogêneas quanto heterogêneas, com diferentes padrões de substituição nas cadeias laterais. Após otimizadas virtualmente quanto a sua energia, as estruturas foram arquivadas para composição de um banco de dados energético-conformacional a ser empregado em aulas práticas de química e bioquímica.
Palavras – chave: Conformação. Configuração. Projeção de Newman. Biomoléculas.
.
Abstract: In order to facilitate learning and minimize difficulties in understanding chemical conformational parameters was proposed to construct a series of virtual carbonic structures, both homogenous as heterogeneous, with different substitution patterns in the side chains. After virtually optimized regarding the energy, the structures were archived to compose an energetical-conformational database useful to be used in practical classes of chemistry and biochemistry.
Key-words: Conformation. Configuration. Newman Projection. Biomolecules.
.
Introdução
O ensino de alguns aspectos químicos e bioquímicos referentes especialmente a configuração e conformação moleculares tem se mostrado bastante complexo e de difícil entendimento por parte dos alunos. A literatura cada vez mais tem apontado a utilização do ambiente computacional para tentar facilitar o entendimento destes aspectos, especialmente porque permite a visualização molecular em três dimensões e, consequentemente, melhora a qualidade do ensino dos aspectos inerentes a configuração e conformação (BARREIRO, 2001; BARREIRO, 1997; FERREIRA, 2008; KLIER, 2012; RAUPP, 2008; RIBEIRO, 2003). Considerando a disponibilidade de inúmeros softwares para ensino de modelagem molecular no mercado, o PcModel mostra-se como uma ferramenta útil e de fácil manipulação, especialmente, para alunos de graduação envolvidos em disciplinas que contemplem conteúdos de química orgânica e bioquímica (SILVA, 2006). A importância do uso dos computadores e das novas tecnologias na educação deve-se hoje não somente ao impacto desta ferramenta na nossa sociedade e às novas exigências sociais e culturais que se impõe, mas também ao surgimento da Tecnologia Educativa (RIBEIRO, 2003; SANTOS, 2001). Eles começaram a ser utilizados no contexto educativo a partir do rompimento com o paradigma tradicional e surgimento do construtivismo, que enfatiza a participação e experimentação do sujeito na construção de seu próprio conhecimento, através de suas interações. Com isso, a capacidade do professor e o conteúdo dos livros constituem uma condição necessária mas não suficiente para garantir a aprendizagem, pois ela envolve um processo de assimilação e construção de conhecimentos e habilidades, de natureza individual e intransferível (FERREIRA, 2008; FREITAS, 1998; RAUPP, 2008; RIBEIRO, 2003; SANTOS, 2001).
Os efeitos do computador na escola dependem de diversos fatores, contudo, a generalidade da investigação aponta para a possibilidade de desenvolvimento de novas competências cognitivas, entre elas: maior responsabilidade dos alunos pelo trabalho, cooperação e novas relações professor-aluno. Assim, o computador constitui-se em uma ferramenta poderosa que pode (e deve) ter todas as suas potencialidades utilizadas com propósitos educacionais, proporcionando ao professor a possibilidade de enriquecer sua prática pedagógica com recursos multimídia, tais como jogos educacionais, vídeos, animações, gráficos e outros materiais que possibilitem ao aluno aprender de forma prazerosa, cativante, divertida e motivadora (RAUPP, 2008). Neste sentido, os jogos educacionais podem ser um elemento catalisador, capaz de contribuir para o “processo de resgate do interesse do aprendiz, na tentativa de melhorar sua vinculação afetiva com as situações de aprendizagem”. A vinculação afetiva exerce um papel fundamental, pois cansado de, muitas vezes, tentar e não alcançar resultados satisfatórios no chamado “tempo” da escola, o aluno experimenta sentimentos de insatisfação constantes, os quais funcionam como bloqueadores nos avanços qualitativos de aprendizagem, além de efetivamente ser utilizado como ferramenta em projetos de pesquisa científica (ANDREI, 2003; BARREIRO, 2001; BARREIRO, 1997; MARQUES, 2010; KLIER, 2012a). Neste sentido, os softwares específicos para modelagem molecular, entre esses o PcModel, constituem importante ferramenta facilitadora de aprendizagem química, haja visto o número de citações enfatizando sua utilização tanto na fundamentação do ensino médio quanto ensino superior. Além disso, o desenvolvimento histórico da química computacional possibilitou que inúmeros softwares fossem desenvolvidos, sendo que um dos seus maiores desenvolvedores, Pople, foi agraciado com o Nobel de Química em 1998, exatamente por sua contribuição ao desenvolvimento de métodos computacionais, o que levou à eventual popularização dos mesmos (FREITAS, 1998). Ainda estamos explorando a potencialidade destes softwares no ensino de química, mas resultados preliminares indicam que existem definitivos ganhos em aprendizagem, que ocorrem devido à formação de uma díade entre o computador e o aluno e da eventual criação de uma percepção cognitiva onde o sistema simbólico da química pode ser manipulado e assimilado pelo estudante. Assim, o computador é utilizado como ferramenta para se adquirir uma “linguagem” química, que se revela potencialmente poderosa para que o estudante seja capaz de resolver situações/problemas químicos. Dessa forma, o uso crescente destes softwares em sala de aula deve ser encorajado, não apenas pelo seu impacto no aprendizado de conceitos e representações químicas e bioquímicas, mas pela eficiência da ferramenta como preparação do estudante para um futuro profissional onde a química computacional estará cada vez mais presente.
.
Objetivo
Utilizando o software PcModel, obter uma tabela comparativa de energias otimizadas para substituintes polares e apolares inseridos em cadeias orgânicas homogêneas e heterogêneas que, quando aplicada em aulas práticas, atendam aos seguintes objetivos específicos:
– facilitar o entendimento de configurações e conformações em projeção de newmann;
– simular rotações em diedros específicos a fim de comparar conformações mais e menos estáveis em estruturas diferentes;
– identificar possibilidades de interações intramoleculares que possam estabilizar uma molécula diminuindo sua energia;
– prever a ordem de grandeza de repulsão eletrônica presente em interações do tipo 1,3-diaxiais;
– associar a estabilidade conformacional e a possibilidade de interação intermolecular aplicada a biomoléculas.
.
Metodologia
Foi empregado o software PcModel para construção molecular de cada uma das cadeias carbônicas homogêneas e heterogêneas, citadas a seguir (figura 1). Obtidas as cadeias, cada uma foi submetida a um padrão de substituição com os substituintes em R1 e R2.
Figura 1 – Cadeias carbônicas (A, B, C e D) e substiuintes propostos (1 a 24)
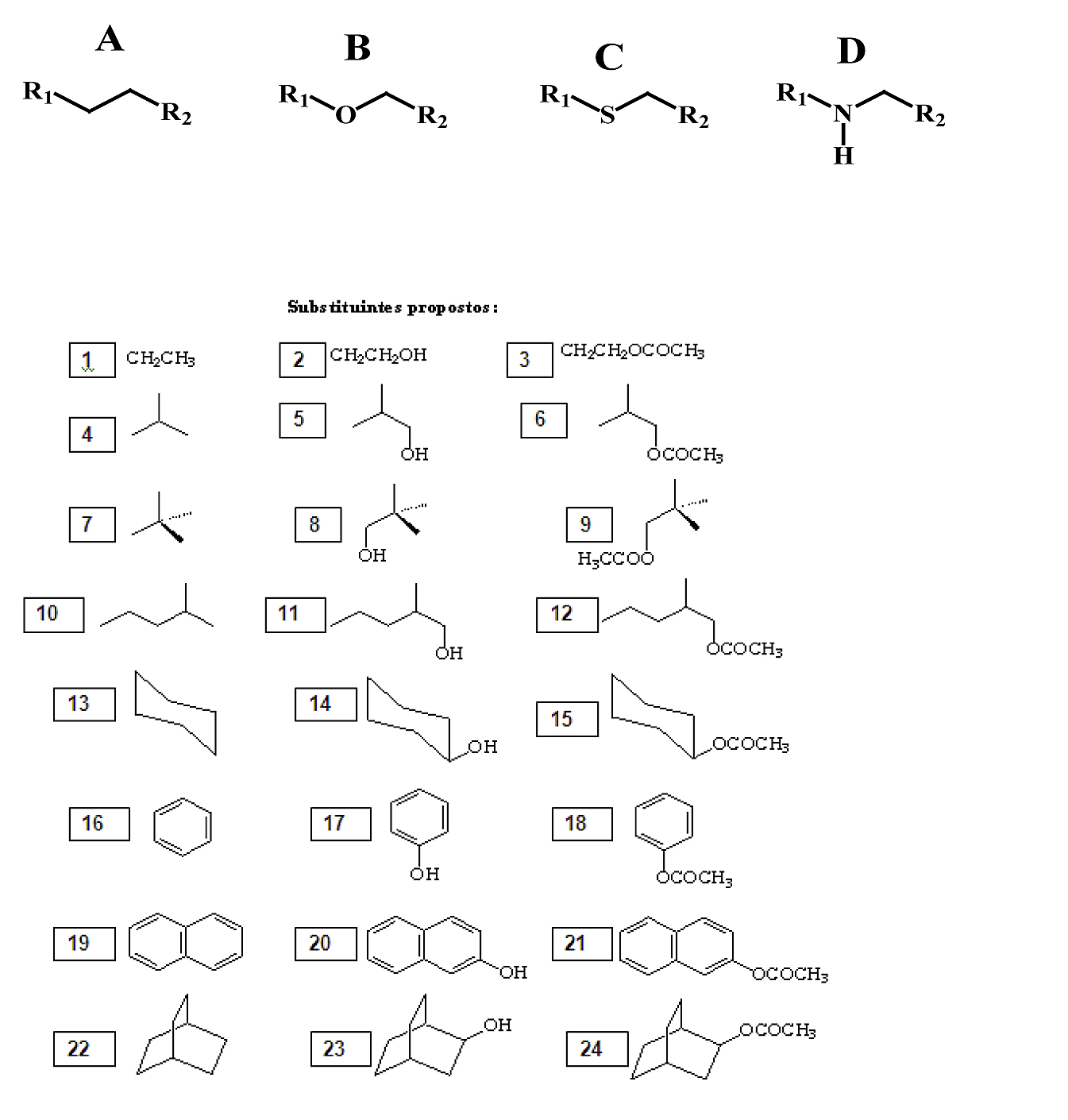
As cadeias propostas contemplam estruturas homogêneas e estruturas heterogêneas oxigenadas, sulfuradas e nitrogenadas, A, B, C e D respectivamente. Os substituintes abrangem oito grupos de substituintes distintos derivados de alcanos, cicloalcanos, aromáticos e bicicloalcanos. Cada grupo de substituintes possui três derivados da mesma estrutura básica, uma não substituída e duas substituídas por hidroxila e carboximetila. Cada uma das cadeias gerou um total de 576 estruturas, considerando os 24 substituintes combinados entre si, totalizando 2304 estruturas distintas que foram analisadas. As projeções de Newmann foram idealizadas ao longo do eixo das ligações C-C na cadeia A, C-O na cadeia B, C-S na cadeia C e C-N na cadeia D. Como banco de dados final, foram gerados 18 valores distintos de energia para cada uma das estruturas geradas, pois consideramos na simulação um giro completo das ligações citadas sobre seu próprio eixo, analisado a cada 20° de giro, o que totalizou 41472 valores de energia tabelados. A utilização do software, bem como a minimização de energia foram feitos segundo dados da literatura (SILVA, 2006; KLIER, 2012; KLIER, 2012a).
Resultados e discussão
As energias minimizadas foram descritas de forma que fosse possível avaliar os substituintes em R1 e R2 de cada cadeia proposta (figura 1), em cada giro parcial de 20° componente do giro completo de 360°, o que resulta em 18 valores de energia para cada estrutura, conforme tabelas 1 e 2.
Tabela 1 – Energias da cadeia A com substituintes 1 a 24 e ângulos de 20° a 180°
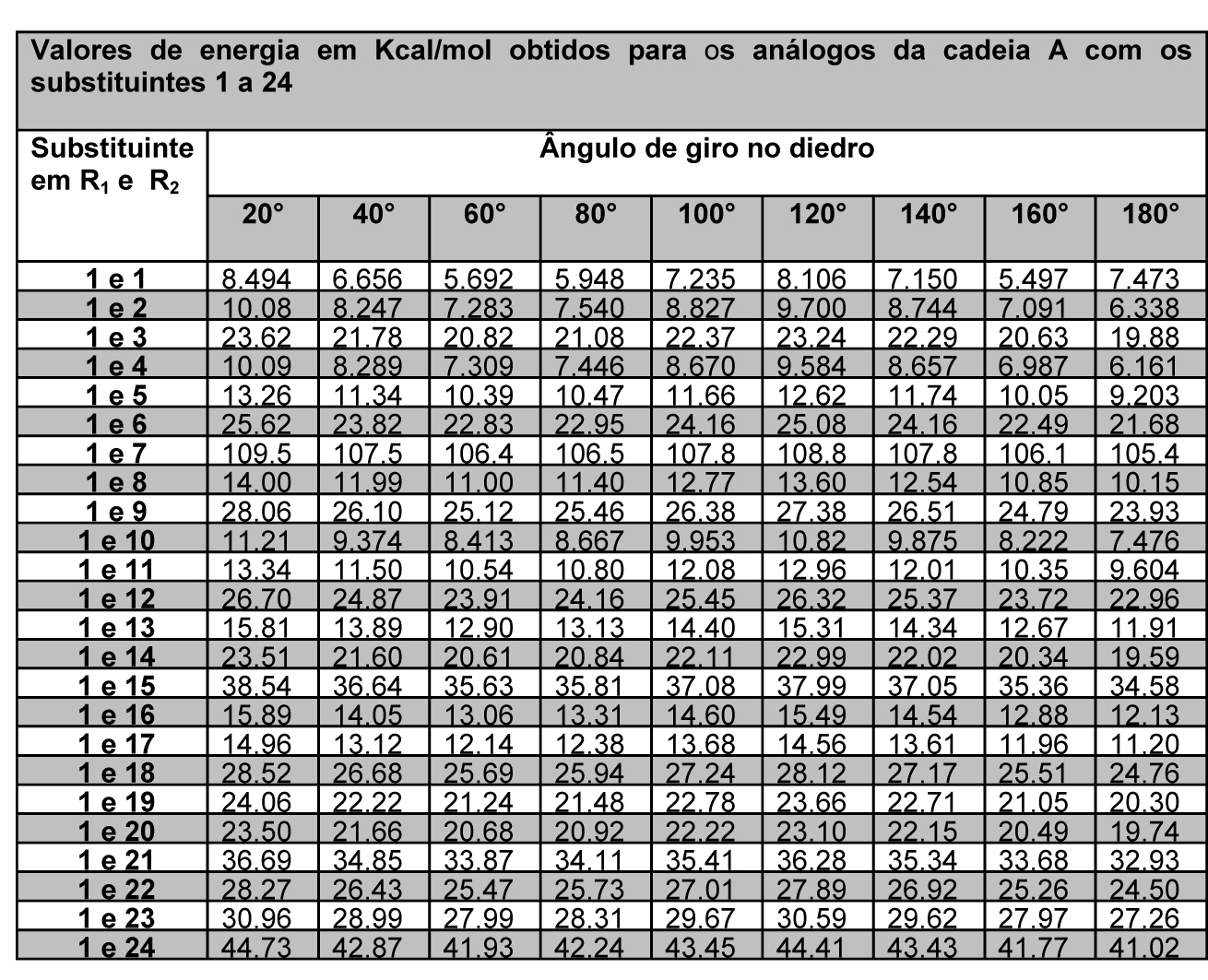
Tabela 2 – Energias da cadeia A com substituintes 1 a 24 e ângulos de 200° a 360°
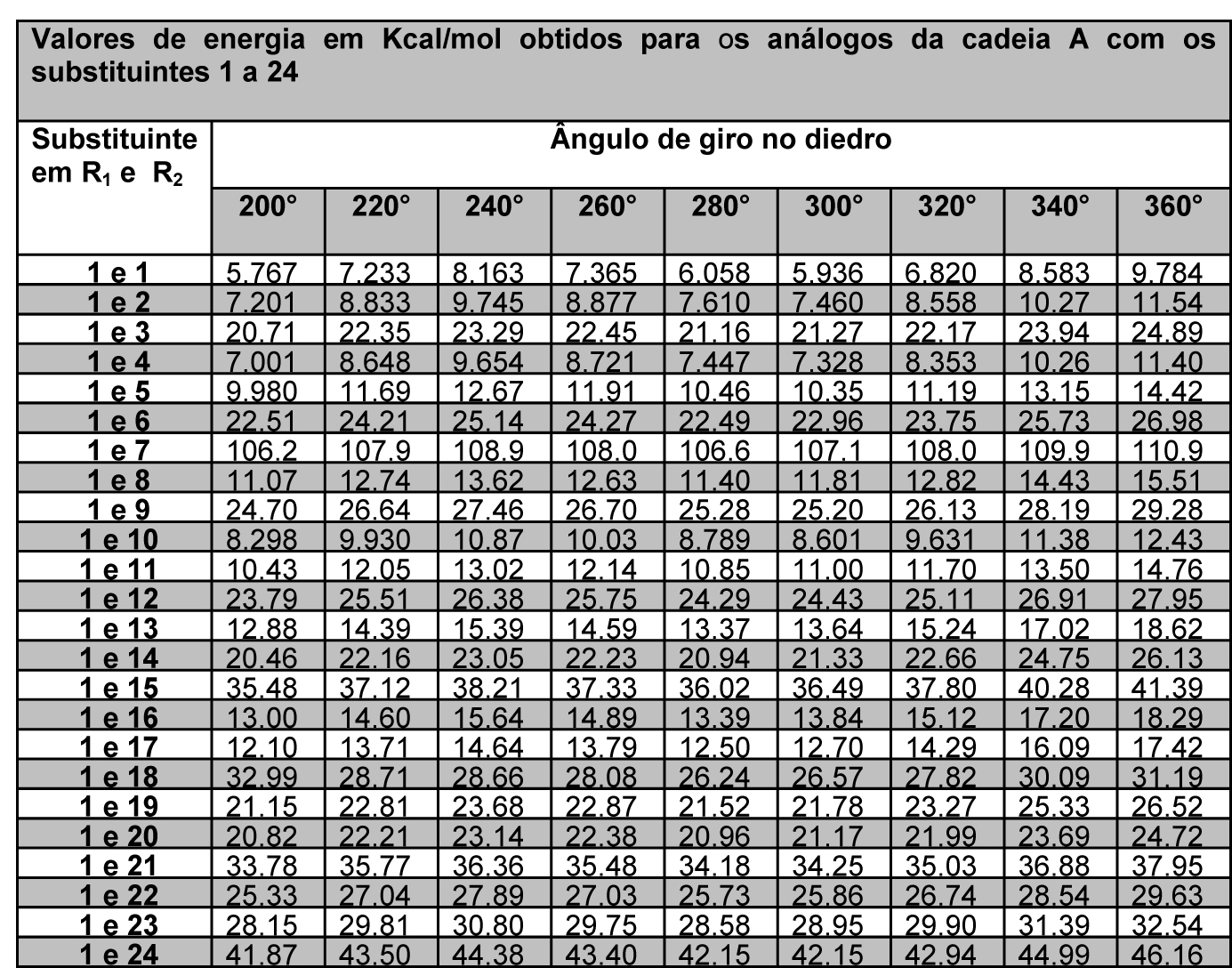
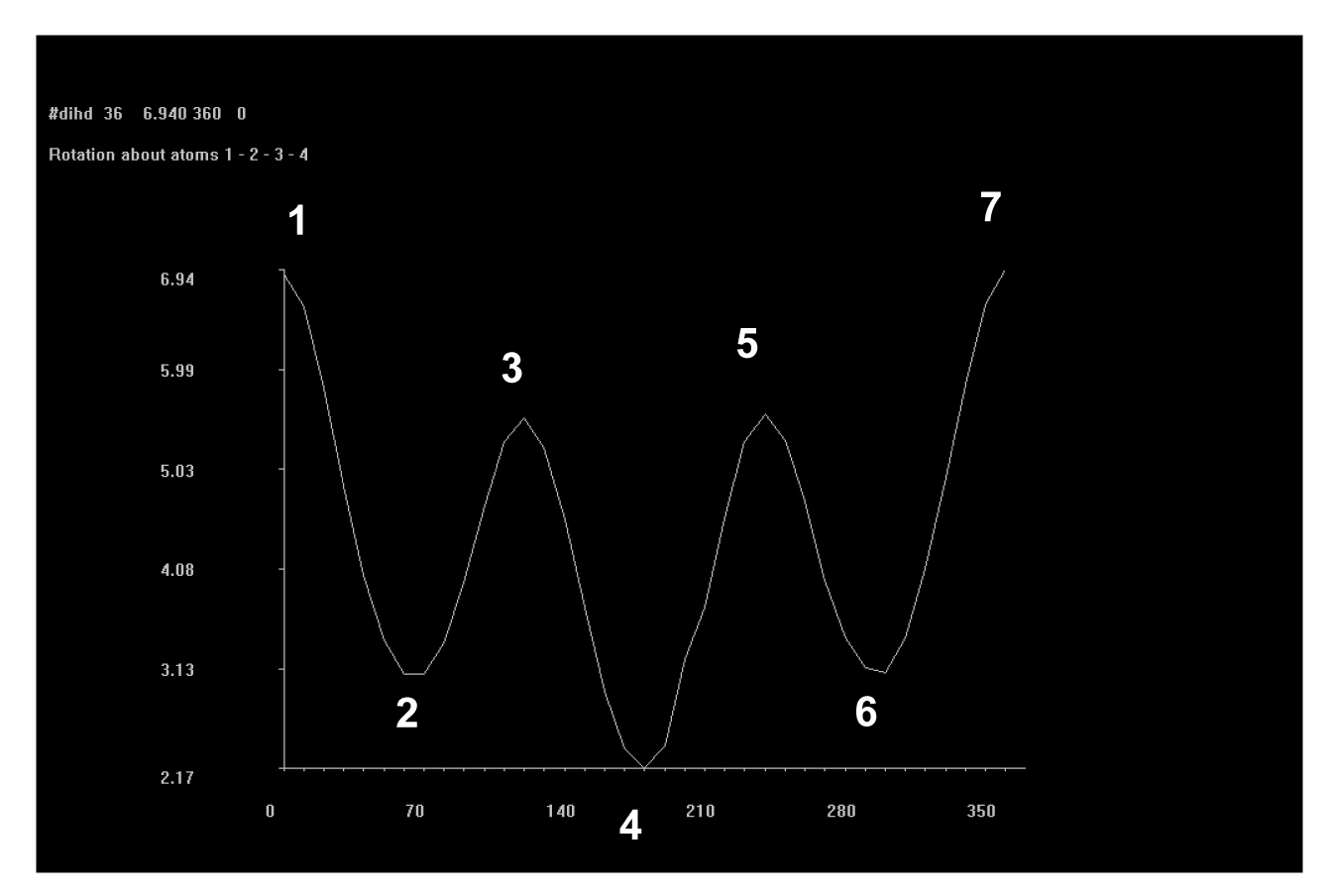
A partir dos dados exemplificados nas tabela 1 e 2, é possível observar que cada cadeia proposta avaliada com os 24 substituintes em R1 e R2 gerou 48 tabelas iguais às exemplificadas, ou seja, um banco de dados final com 192 tabelas. Com a definição dos substituintes variáveis quanto a sua massa molar, foi possível estabelecer um padrão de estabilidade dentre as estruturas avaliadas, considerando que são possíveis quatro conformações padrão nas projeções de newmann a partir do ângulo de giro entre os substituintes – sinperiplanar, sinclinal, anticlinal e antiperiplanar. Estas conformações são obtidas em momentos distintos no giro do ângulo diedro em cada cadeia proposta, conforme observado na figura 2, que apresenta um modelo gráfico obtido no PcModel para o butano. Observa-se que os pontos identificados 1 e 7 referem-se à conformação sinperiplanar, com proximidade máxima entre os substituintes, sendo 0° ou 360°, os pontos 2 e 6 referem-se à conformação sinclinal, com 60° e 300° de giro, os pontos 3 e 5 referem-se à conformação anticlinal, com 120° e 240° de giro, e o ponto 4 refere-se à conformação antiperiplanar, com 180° de giro e distância máxima entre os substituintes.
Figura 2 – “Print screen” da tela do software PcModel localizando as conformações em projeção de newmann segundo ângulo de giro
Tomando como exemplo as conformações geradas pelo substituinte 1 em R1 (figura 1) combinado com os 24 substituintes possíveis em R2, sempre na conformação de maior repulsão eletrônica entre os mesmos, conformação sinperiplanar, podemos obter um perfil de correlação de energia e massa molar dos substituintes no gráfico 1.
Gráfico 1 – Energias das conformações geradas pelo substituinte 1 em R1 e os substituintes 1 a 24 em R2
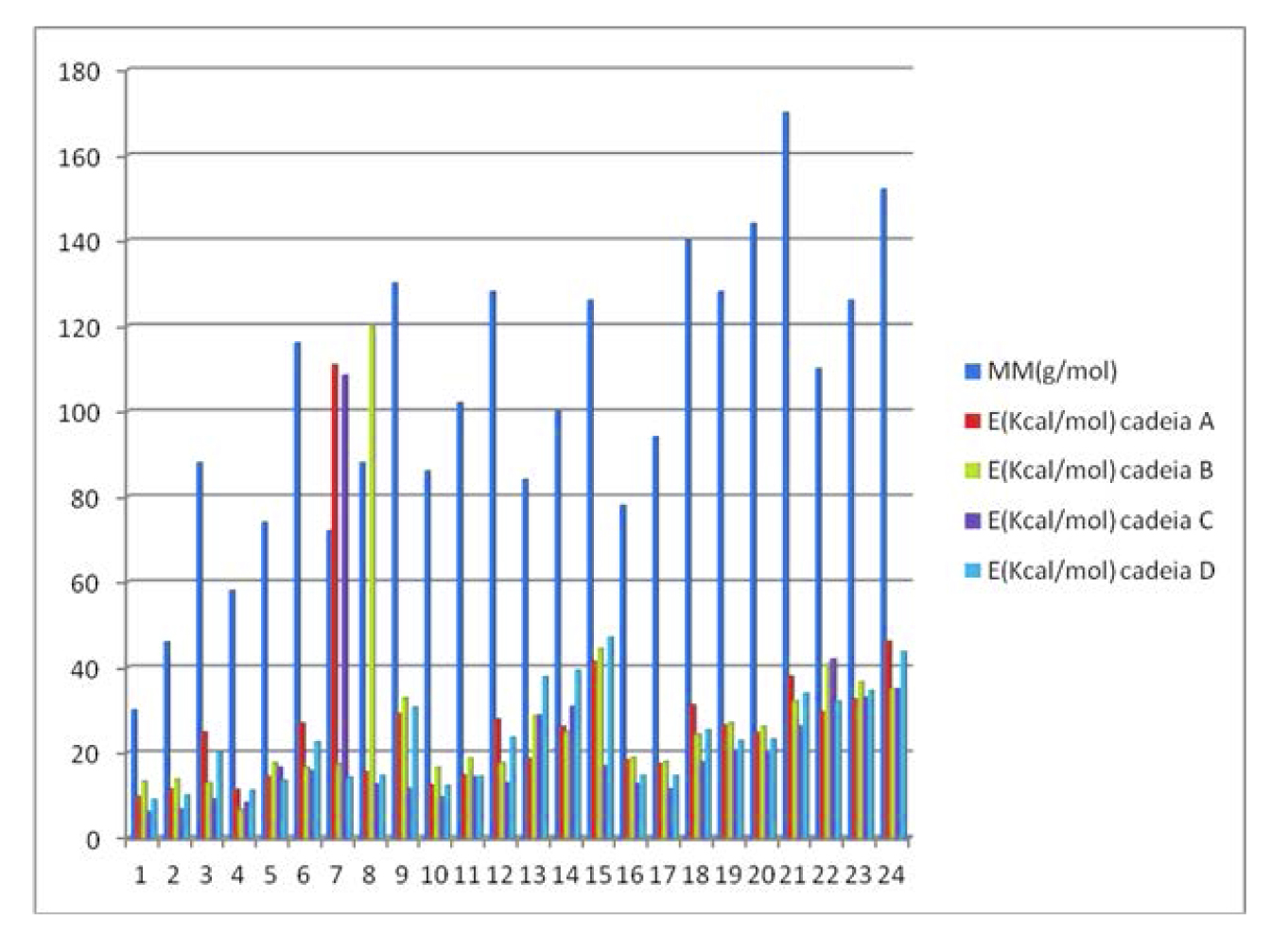
Ao avaliarmos os padrões de variação da energia total dentre os compostos obtidos, é possível observar um padrão homogêneo de variação crescente, quando confrontamos as energias dentro dos oito patamares crescentes de massa molar, exceto nos substituintes de 7 a 9. Ou seja, como temos oito padrões estruturais de substituintes, com três variações constantes e crescentes de massa molar em cada um deles, era esperado que numa condição conformacional antiperiplanar as interações repulsivas entre grupos volumosos elevassem os padrões de energia como foi observado. Porém, nos substituintes 7, 8 e 9, estas variações de energia foram bem superiores aos demais padrões moleculares, possivelmente pela existência de um carbono quaternário na estrutura do substituinte. Este aumenta consideravelmente a compressão estérica e, consequentemente, a repulsão eletrônica envolvida no substituinte, especialmente em função dos rotâmeros possíveis em uma mesma conformação, como pode ser observado na figura 3.
Figura 3 – Rotâmeros da conformação sinperiplanar
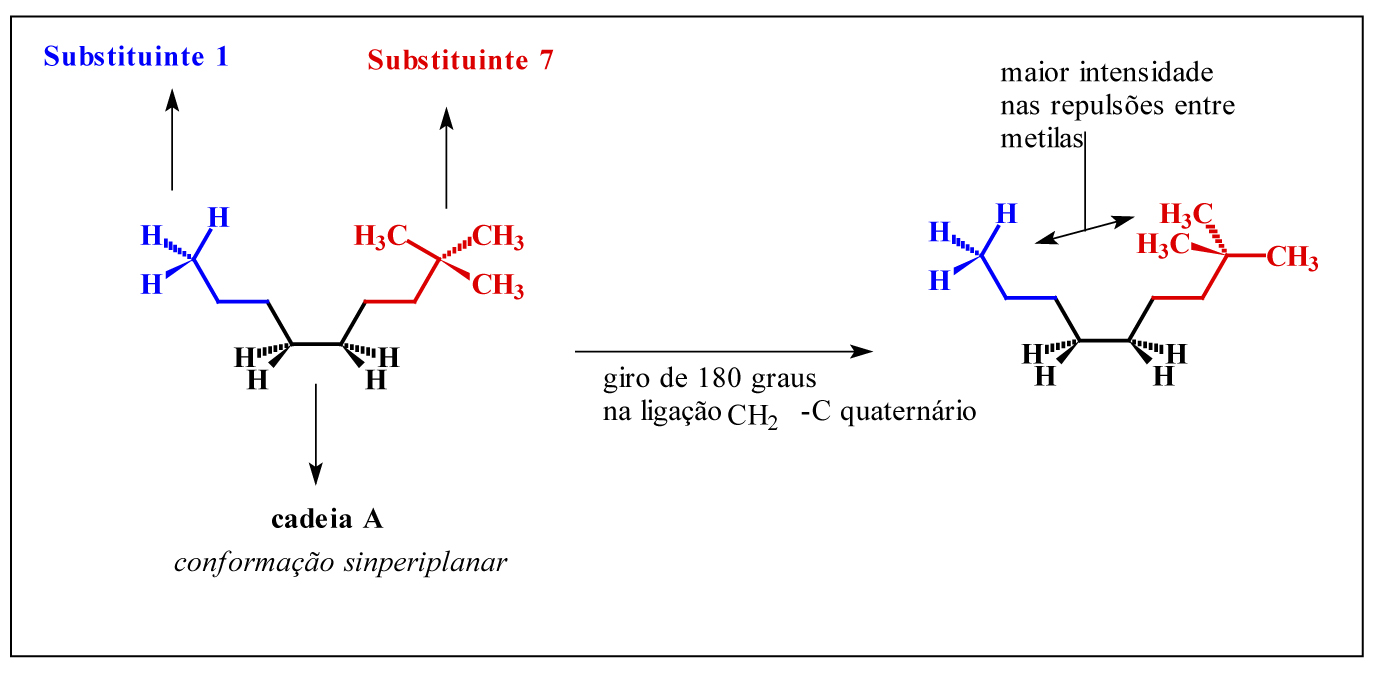
Como pode ser observado na figura 3, um giro de 180 graus na ligação metileno-carbono quaternário do substituinte 7, aumenta significativamente a repulsão eletrônica entre os grupamentos vizinhos, o que contribui para uma variação ainda maior de energia nestas estruturas, uma vez que com as ligações entre carbonos vizinhos eclipsadas, a conformação sinperiplanar possui maior energia total.
A fim de avaliarmos a influência estrutural da inserção de heteroátomos nas cadeias propostas, avaliamos as quatro cadeias com substituintes iguais em R1 e R2 na conformação mais estável antiperiplanar, numa conformação estrutural que podemos considerar simétrica quanto à vizinhança nos carbonos substituídos. Os resultados de energia obtidos para cada uma das cadeias pode ser observado no gráfico 2. Mesmo na conformação antiperiplanar simétrica, tanto os substituintes 7 e 9 como os substituintes 17 a 19 apresentaram substancial aumento da energia da estrutura final gerada. Os substituintes 7 e 9 apresentaram maiores variações nas cadeias heterocíclicas, B, C e D, lembrando que em estruturas simétricas, tanto R1 quanto R2, estarão substituídos com grupamentos quaternários sendo um deles contendo o grupo funcional carboximetila. Entretanto, não se observou aumentos significativos na energia das cadeias com o substituinte 8, também quaternário funcionalizado com o grupo funcional hidroxila. Tal comportamento provavelmente seja devido à existência de interação intramolecular por ligação de hidrogênio entre a função hidroxila; doadora de interação por ligação de hidrogênio, e os elétrons livres do heteroátomo; aceptor de ligação de hidrogênio, interação esta que estabiliza a molécula diminuindo sua energia total.
Já os substituintes 17, 18 e 19 correspondem respectivamente aos grupamentos para-hidroxifenil, para-carboximetilfenil e naftil. Mesmo em uma conformação teoricamente favorável estericamente, é possível observar que tais grupamentos são capazes de alterar
Gráfico 2 – Energias das cadeias propostas na conformação sinperiplanar simétrica
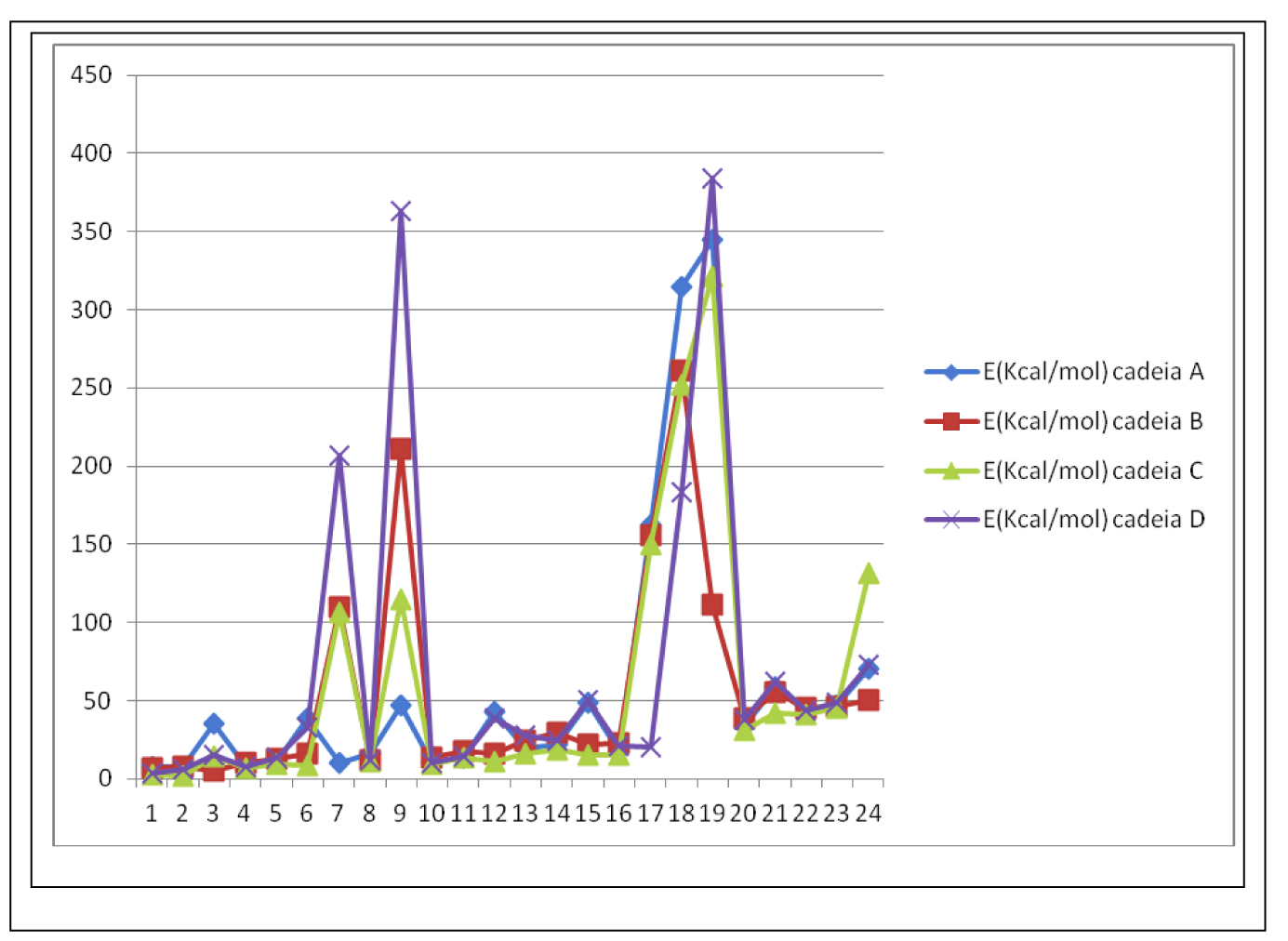
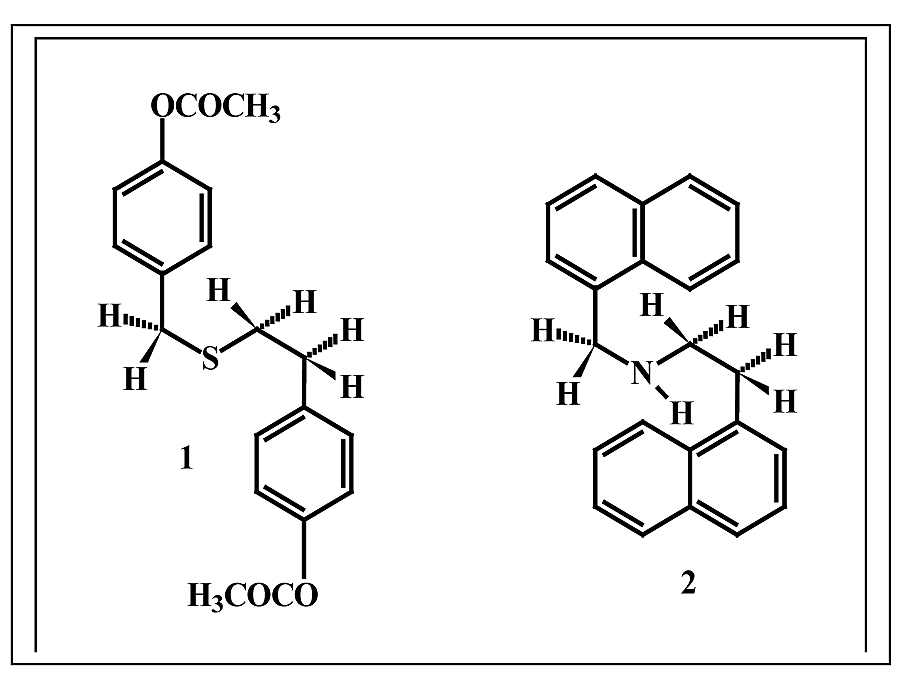
drasticamente o perfil energético-molecular sem distinção da cadeia principal. Tal variação se deve também mais provavelmente à existência de rotâmeros, que aproximam as cadeias laterais aumentando a repulsão eletrônica, especialmente, nos substituintes 18 e 19, gráfico 2. Porém, tais rotâmeros não são observados nos substituintes 20, 21, 22, 23 e 24. Possivelmente, o fato de serem planos, sistema monoaromático-paracarboximetil funcionalizado no substituinte 18 e sistema biaromático não funcionalizado no substituinte 19, sejam os “limites” de volume molar que possibilitem giros livres dos rotâmeros em relação à cadeia principal promovendo variação de energia total, conforme figura 4.
Figura 4 – Rotâmero da cadeia C com substituinte 18 (1) e rotâmero da cadeia D substituinte 19 (2)
.
Conclusão
Um banco de dados com as cadeias homogêneas e heterogêneas foi obtido a partir dos dados gerados pelo software. Tais informações complementam o entendimento da química e podem ser empregadas para o estudo da mesma. Formas de interação e ensino de conformações moleculares, aproveitando-se de recursos tecnológicos são estimuladas.
.
Referências
ANDREI, C.C., FERREIRA, D.T., FACCIONE, M., FARIA, T.J. Da Química Medicinal à Química Combinatória e Modelagem Molecular: um curso prático. Barueri, SP: Manole, 2003. 154p.
.
BARREIRO, E.J. Sobre a química dos remédios dos fármacos e dos medicamentos. Quim. Nova na escola, n.3, 2001.
..
BARREIRO, E.J., RODRIGUES, C.R., ALBUQUERQUE, M.G., RABELLO DE SANT’ANNA, C.M., ALENCASTRO, R.B. Modelagem Molecular: Uma Ferramenta para o Planejamento Racional de Fármacos em Química Medicinal. Quim. Nova, v.20, n.1, 1997.
.
FERREIRA, P.F.M., JUSTI, R.S. Modelagem e o “Fazer Ciência”. Quim. Nova na escola, n.28, 2008.
.
FREITAS, L.C. Prêmio Nobel de Química 1998. Quím. Nova na Escola, n. 8, 6, 1998.
.
KLIER, A.H. Conformações do cicloexano: um modelo de estudo no PcModel. Pós em Revista, n. 5, 2012.
.
KLIER, A.H., SABINO, G.S., LEAL, S.C., PEREIRA, A.F.A., MAPA, L.A., FERREIRA, L.E.C., MOREIRA, N.M., CHIES, P.G. Diedros conformacionais e sua aplicação no estudo de estabilidade de biomoléculas. Pós em Revista, n. 6, 2012a.
.
MARQUES, M.V., RUSSOWSKY, D., FONTOURA, L.A.M. Análise Conformacional de Compostos de Biginelli com Atividade Antineoplásica. Eclet. Química, v.35, n.4, 2010.
.
RAUPP, D., SERRANO, A., MARTINS, T.L.C. A evolução da química computacional e sua contribuição para a educação em química. Revista Liberato, v.9, n.12, 2008.
.
RIBEIRO, A.A.; GRECA, I.M. Simulações Computacionais e ferramentas de modelização em educação química: uma revisão da literatura publicada. Quím. Nova, , v. 26, n. 4, 2003.
.
SANTOS, H.F. O conceito da modelagem molecular. Quim. Nova na escola, n.4, 2001.
.
SILVA, T.H.A. Practica III.3 Modelagem molecular com o auxílio do computador, 2006. Disponível em http://old.iupac.org/publications/cd/medicinal chemistry/Practica-III-3.pdf
.
Notas
1-Discentes do curso de Farmácia, alunos de Iniciação Científica do Centro Universitário Newton Paiva.
2-Colaboradores da pesquisa, Docentes do Centro Universitário Newton Paiva.
3-Coordenador da pesquisa, Docente do Centro Universitário Newton Paiva.